LEDGF/p75
Integrase Inhibitors and Capsid Assembly Inhibitors Offer New Approaches
for Blocking HIV Replication
 |
 |
 |
 |
 |
 |
 |
| SUMMARY:
While the drug development pipeline is not as full as it has
been in recent years, researchers continue to work on new
approaches to antiretroviral therapy. Two such novel approaches
-- LEDGF integrase inhibitors and capsid assembly inhibitors
-- were described in oral presentations at the recent 17th
Conference on Retroviruses and Opportunistic Infections (CROI
2010) last month in San Francisco. |
|
 |
 |
 |
 |
 |
 |
 |
By
Liz Highleyman
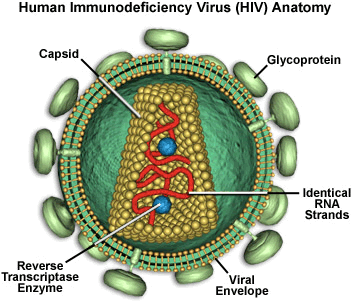
In order
to replicate, HIV must enter a
host cell, shed its outer coat, copy its genetic material (transcribing
RNA into DNA, the job of reverse transcriptase), and insert the new
copy into the host cell's chromosome (the job of integrase). The virus
then "hijacks" the cell's machinery to produce large viral
proteins, which are cut up (the job of protease) and assembled into
new viral particles.
LEDGF
Inhibitors
Frauke Christ from Catholic University in Leuven, Belgium, presented
data on a new type of HIV integrase inhibitor that interferes with a
cellular protein known as LEDGF (lens epithelium-derived growth factor,
or p75). Unlike the approved integrase inhibitor raltegravir
(Isentress), this approach does not interfere with the integrase
enzyme directly, but rather focuses on its "binding partner,"
LEDGF/p75.
In 2003, the investigators identified LEDGF/p75 as a strong integrase
binding partner, and they have since established a validation procedure
for novel HIV cofactors. This allowed them to confirm the importance
of LEDGF/p75 -- which tethers HIV integrase to the cellular genome --
in the process of HIV replication.
Understanding another step in the HIV lifecycle, of course, offers a
new drug target. X-ray crystallography of the integrase-binding domain
revealed that small molecule protein-protein interaction inhibitors
could potentially disrupt the LEDGF/p75-integrase interaction.
The researchers used rational drug design to identify small molecules
likely to fit the interaction site, narrowing their search to a set
of compounds known as 2-(quinolin-3-yl) acetic acid derivatives (dubbed
LEDGINs). From an initial 2 dozen candidates, they found one (CX00287)
that moderately inhibited LEDGF/p75-integrase interaction and HIV replication
in vitro.
They tinkered further with the shape of most promising candidate, creating
optimized versions. The most promising, CX04328, was a potent inhibitor
of LEDGF/p75-integrase interaction and HIV replication -- but not of
other cellular functions -- in laboratory studies. It showed no toxicity
in cell cultures and was not cross-resistant with strand-transfer integrase
inhibitors such as raltegravir and the experimental integrase inhibitor
elvitegravir.
The selectivity of the chosen compound was considered inadequate, so
work continued, yielding a modified compound -- CX06387 -- that was
the most potent so far, was highly selectivity, and produced good preliminary
toxicity data, making it a promising candidate for further clinical
development. Christ said initial animal studies are underway and projected
that these might be finished in about a year.
"Our work demonstrates the feasibility of rational design of small
molecules inhibiting the protein-protein interaction between a viral
protein and a cellular host factor," the investigators concluded.
The discovery of the 2-(quinolin-3-yl) acetic acid derivatives as the
first agents to inhibit the LEDGF/p75-integrase interaction and HIV
replication "provides the ultimate proof for the crucial role of
the co-factor in HIV replication."
Capsid Assembly Inhibitors
Steve
Titolo from Boehringer Ingelheim Canada and colleagues described the
development of novel capsid assembly inhibitors, which prevent construction
of the cone-shaped shell surrounding newly produced viral genetic material
-- a crucial step in forming functional new viral particles. There are
currently no approved assembly inhibitor dugs.
The investigators developed an in vitro capsid assembly assay,
which they used to screen libraries of potential compounds. Inhibition
of wild-type and drug- resistant virus was studied in C8166 T-cells.
Binding to the capsid was determined using nuclear magnetic resonance
spectroscopy and X-ray crystallography.
Screening yielded several different clusters of structurally related
compounds (dubbed chemotypes) that inhibited capsid assembly. The researchers
optimized 2 promising chemotypes -- benzodiazepines and benzimidazoles
-- producing compounds with potent antiviral activity against both wild-type
HIV and virus resistant to existing antiretroviral drug classes.
The two
chemotypes both reduced production of infectious virions (virus particles),
but they had different effects, with benzodiazepines preventing formation
of mature virions and benzimidazoles causing production of progeny virus
with profound core morphology defects.
Structural
analysis showed that both selected chemotypes bound to the N-terminal
domain of capsid by forming a binding pocket, which overlaps with the
binding site for a previously reported type of capsid assembly inhibitors.
However, the 2 chemotypes had different shapes, with the benzimidazoles
protruding more from the binding pocket.
Letting
the virus replicate in the presence of the new inhibitors led to emergence
of several resistance mutations, mostly in highly conserved regions
in or near the binding pocket; different sets of resistance mutations
were selected by the 2 chemotypes, but there was some cross-resistance.
Most of the resistance mutations impaired viral replicative capacity.
Development
of the 2 specific lead compounds was discontinued due to potency issues,
which Titolo said was probably due to their lipophilic nature.
Nevertheless, the researchers concluded that their work "demonstrated
a proof of concept" that potent capsid inhibitors could be developed
as new anti-HIV drugs.
Study 1: Katholieke Univ Leuven, Belgium;
Center for Innovation and Stimulation of Medical Development, Leuven,
Belgium.
Study 2: Boehringer-Ingelheim Ltd, Laval,
Canada; University of Utah, Salt Lake City, UT.
3/2/10
References
F Christ,
A Voet, A Marchand, and others. First-in-class Inhibitors of LEDGF/p75-integrase
Interaction and HIV replication. 17th Conference on Retroviruses &
Opportunistic Infections (CROI 2010). San Francisco. February 16-19,
2010. Abstract 49.
S Titolo,
J-F Mercie, E Wardrop, and others. Discovery of Potent HIV-1 Capsid
Assembly Inhibitors. 17th Conference on Retroviruses & Opportunistic
Infections (CROI 2010). San Francisco. February 16-19, 2010. Abstract
50.

